| 11 混合エントロピーと気体の化学ポテンシャル | ||
| f-denshi.com [目次へ] 更新日: 21/10/24 | ||
| サイト検索 | ||
前ページ10.では化学ポテンシャルの一般論からギブス-デュエムの式などを導きました。しかし,残念ながらそこで導いた公式たちは,それだけではほとんど無力で,活躍の場が限定されてしまいます。それらを最強の軍団に仕立て上げるために必要なのが,「混合エントロピー」です。化学ポテンシャルの化学への応用のためには,混合エントロピーを用いた,「多成分系の化学ポテンシャルの具体的表式」が必要なのです。
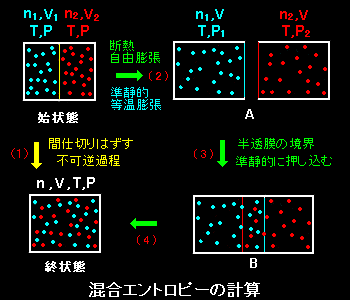
[1] 2種類の理想気体とみなせる n1モルの気体1と n2モルの気体2が,間仕切りで体積が V1,V2 に分離された断熱容器の中にそれぞれ左右分かれて閉じ込められており,同じ温度T,圧力P にあるとします(始状態)。ここで,
(1) 間仕切りを取り除くと,2種類の理想気体は均一に混じりあい,平衡状態に達する(終状態)。このときの温度,圧力は,T,Pのままで,粒子数はn=n1+n2,体積はV=V1+V2となる。
この現象はもちろん不可逆過程ですが,系は外部と熱や仕事の交換をまったくせず,エントロピー変化だけが起こります。
このように断熱容器の中で理想気体とみなせる2種類の気体を混合する時のエントロピー変化を混合エントロピーといいます。
[2] この過程は不可逆過程なのでエントロピーを直接計算できません。計算を可能にするためには,始状態と終状態とを結びつける準静的な過程を見つける必要があります [#] が,そこで考え出されたのが半透膜を使う次のような2種類の気体の混合方法 (思考実験) です。
まず,始状態にある気体をそれぞれ別々の異なる容器の中で,
(2) 断熱自由膨張によってそれぞれの気体の体積をともに,V1+V2 にする。
この過程そのものは不可逆ですが,後ほど述べるように同じ状態変化を与える可逆的な等温膨張過程に置き換えることが可能です。ここで,この容積がV1+V2である断熱容器は半透膜でできており,
気体1を閉じ込めている容器は気体1は全く通さないが気体2は自由に通す。
気体2を閉じ込めている容器は気体2は全く通さないが気体1は自由に通す。
という性質を持っているとします。次に,
(3)半透膜の容器を互いに入れ子の状態に押し込んでゆくと,容器の重なった部分には両方の気体が入ります。
(4)重なりが完全になるまでゆっくり押し込むと,体積 V1+V2 の中で2種類の気体も完全に混合されます。(終状態)
このような半透膜を利用した気体の混合過程をすべて準静的な過程で行えば,,エントロピーの計算が可能です。
[3] それでは計算です。
(2)の断熱(自由)膨張過程は不可逆過程なので,そのまま熱の出入りが 0 であることを用いてエントロピーの計算をすることはできません[#]。
(つまり,エントロピー変化が 0 とするのは間違い!教科書にはふつう正しいことだけが記述されがちだが,間違いを示して,それがなぜ間違いかを理解することも重要だと思う。)
しかし,理想気体はこの過程で温度変化がないので,これを可逆的な等温膨張過程に置き換えて,エントロピー変化を計算することができます。
「不可逆過程のエントロピー変化の計算 ⇒ その過程と同一の始状態,終状態をもつ可逆変化を探せ!」
ということでした。
熱力学第一法則 [#] においてdU=0とすると,TdS=PdV。これに理想気体の状態方程式を用いて,
dS= PdV = nR dV T V
この関係式より,準静的等温膨張による気体1のエントロピー変化は,
| ΔS1= | V1+V2 | n1R dV | =n1R log | V1+V2 | ≒n1R log | n1+n2 | =−n1R log x1 | |
| V1 | V | V1 | n1 |
と計算できます。ここで,≒のところでドルトンの分圧の法則[#]を用いています。同様に,
ΔS2=−n2R log x2
となります。半透膜を押し込む準静的断熱過程(各々の気体についてみると何事も起こっていないでしょ!)ではエントロピー変化は 0 なので,結局,いま計算したΔS1とΔS2の和が始状態と終状態とのエントロピー差になります。すなわち,
ΔSmix=ΔS1+ΔS2
=−R (n1log x1+n2log x2) [混合エントロピー]
となります。ここで,0<x1,x2<1 であることから,必ず,ΔSmix>0 であることにも注意しましょう。
エントロピーはもともと,熱の移動に着目して定義された物理量でした。しかし,ここでの考察は,「熱の移動が全くないような状況においてエントロピーが変化(増大)することがある。」 ことを示唆しています。ケルビンらによって完全に捉えられたかのようなエントロピーも,実はその真の姿が映し出す影を追っているだけに過ぎないのではないかと混合エントロピーが語りかけてきます。この声を聞き漏らさなかった人物こそが,ボルツマン なのですが,ここではこれ以上深入りしません。
混合エントロピーに関する「ギブスのパラドックス」について欄外に説明があります。⇒ [ギブスのパラドックス]
[4] さて,これより混合気体のエントロピーは,純粋な気体1,2 のモルエントロピーをそれぞれ,s10,s20 として [ 注意 ] ,
S=n1s10+n2s20−R ( n1 log x1+n2log x2)
=n1{ s10−R log x1}+n2{ s20−R log x2}
と書くことができます。理想気体の混合前後でエンタルピーは不変で,純粋な気体1,2 の1モルあたりの量をそれぞれ h10,h20 とすれば,
H=n1h10+n2h20
と単純な和で表せます。したがって,混合気体のギブスの自由エネルギー G =H−TS は,
G=n1h10+n2h20−T[ n1{s10−R log x1}+n2{s20−R log x2} ]
=n1{ h10−Ts10+RT log x1 } + n2{ h20−Ts20+RT log x2 }
と書いて構わないでしょう。
純物質について,モル自由エネルギーと部分モル自由エネルギーは一致するすで [#],
h10−Ts10=g10=μ10
h20−Ts20=g20=μ20
と書くこともできます。
[5] 以上,まとめると,
|
混合(理想)気体のギブス自由エネルギーと化学ポテンシャル G=n1μ1+n2μ2 ただし,μj0=hj0−Tsj0 [純粋な j 成分気体1mol のギブス自由エネルギー]。と書き表すことができ,標準化学ポテンシャルと呼ぶ。また, を 混合気体の成分 j の化学ポテンシャル という。 ( j = 1,2 ) |
この議論は成分がm個に増えてもそのまま拡張できるのは説明するまでもないでしょう。気体からなる多成分系のギブス自由エネルギーの具体的な形は,
G = njμj ; μj=μj0+RT log xj ,xj=nj /(n1+・・・+nm)
理想気体とみなせる気体どおしを混合した多成分系では,気体成分の一つ一つの化学ポテンシャルはその成分の純粋状態での化学ポテンシャルとその成分のモル分率とに分離された式で表されるのです。化学ポテンシャルを,
温度,圧力依存性と組成依存性とを分離して考えることができる
といってもよいでしょう。厳密に言えば,そのように取り扱うことのできるモデルをここに提示したということになりますが,このように各成分ごとに具体的に化学ポテンシャルを分離した形で書き下すことで,多成分系の熱力学的取り扱いを個々の成分からの寄与の寄せ集めとして捉えたり,熱力学状態を相対的な比較から絶対的な基準を定めて,定量的,体形的に取り扱いことが可能となります。
補足
化学ポテンシャルが基準となる圧力P0を用いて,
μ(T,P)=μ0(T,P0)+RTlog P P0
で与えられることを通して混合エントロピーを求める方法もあります。
複数の方々(学生さん)から同じような質問を度々頂いたこともあり,簡単な説明をここに書いておくことにしました。 (2008/9/24)
ギブスのパラドックスとは?
本文中では,それぞれ,n1,n2モルの2種類の気体を混合したときのエントロピー変化が,
(A) ΔSmix=−R(n1 log x1+n2log x2) > 0 [混合エントロピー]
であることを示しました(記号の意味は本文中と同じ)。
ところが,間仕切りの左右の気体1と気体2がもし同じ温度,圧力にある同種類の気体だとしたら,間仕切りを取り去っても巨視的には何も変化は起こりません。したがって,エントロピーも変化しないはずで,
(B) ΔSmix=0
でなければならないという主張もできるはずです。
本文中での混合エントロピー(A)を導出する際には,気体1,気体2の個性を示すような物理量は何も使ってないので,一見,同一種類の気体の混合に対しても(A)のような計算ができるように見えます。よって,(A)と(B) の2通りの結果が導かれるのは矛盾ではないかというのが(熱力学における)ギブスのパラドックスと呼ばれるものです。
まず,結論として,同一種類の気体の混合に対して,どちらの混合エントロピーが正しいかと言えば,(B)です。同一種類の気体の混合によるエントロピー変化はゼロです。さらに,同一粒子に対しても,(A)のようにエントロピーが計算できるとの主張が誤りであることは,少し考えれば以下に示す通り容易に理解できます。
では,同一の粒子に対して,(A)の方法で混合エントロピーを計算することがなぜ誤りであるかを示したいと思います。
(A)の計算方法と(B)の計算?方法で何が違うのかと言えば,
(A) 左右にある同一種類の粒子を区別できるものとして扱っている。
(B) 左右にある同一種類の粒子は区別できないものとして扱っている。
という違いがあります。つまり,(A)においては,容器の間仕切りの左側にあった気体1も右側にあった気体2も,間仕切りを取り去った後には容器全体に広がるとして,エントロピー変化を計算していることになります。つまり,もともと粒子が左側にあったのか右側にあったかが識別可能として,混合によるエントロピー変化を計算していることになります。もし,左右に同一ではなく異なる種類の気体が入っていたならば,たしかに混合後も気体粒子の一つ取り出したとき,それが元々,右側にあったか左側にあったか言い当てることができます。しかし,左右に同一種類の気体が入っていた場合はそれができず,(A)の処方箋は無効となるのです。
(A)の処方箋が無効となる理由は,本文中の過程(3)で用いる半透膜を仮想的にも用意できないことにあります。なぜならば,「気体1は通過させるが,気体2は通過させない半透膜・・・」 という記述は,もし,両側ともに気体1であったとすると,「左容器内の気体1は通過させるが,右容器内の気体1は通過させない半透膜・・・」 と言い換えなければなりません。しかし,こんな半透膜は存在しません。
なぜならばこれが可能とすると,次のような実験が可能となります。すなわち,容器左右全体に気体1が入った状態で,その中央にこの膜を設置します。すると,気体1は左から右へしか移動できませんから,十分な時間放置すれば,気体1をすべて容器の右側に集めることができます。もちろん,このとき,外界には何の変化も起きません。これは熱力学第2法則に反することです。
すると,本文中の混合エントロピーの算出方法が胡散臭い,根拠が曖昧(=ギブスのパラドクス)という話が正統性をもつならば,熱力学第2法則そのものが間違っているということになってしまいます。したがって,熱力学第2法則が正しいという立場であれば,熱力学におけるギブスのパラドクスは存在しないということです。
一方,(B)の間仕切りを取り払った後に何も起きないという主張には,容器の中を動きまわる同一種類の粒子を一つ一つ区別することは不可能だということが暗に含まれています。この前提があるからこそ間仕切りをとっても何も起きないと主張できるわけです。もし,間仕切りを取り除いてしばらくしてからも,「はじめ右にあった粒子が左に移動しよった」というようなことがわかるのなら,それは,「間仕切りをとっても何も起きない」という主張とは相反します。つまり,(A)と(B)とでの混合エントロピーの解釈の違いは同じ体積内を動き回る同一種類の粒子の識別可能性の可否に基づいて生じているのです。
「マクスウェルの悪魔」で検索して調べてみると,この問題に関連する議論が永遠と1980年ごろまで(もしかしたら現在までも)続いていたことが分かります。
もちろん,このパラドクスは単に(修正古典)統計力学を持ち出せば解決するという話なんかでもありません。(統計力学においてもギブスのパラドックスを登場させる人たちも存在します。)
量子力学では,多粒子系の波動関数として,同種粒子の交換に対して,波動関数が不変[対称](ボース粒子)であるか,符号が反転する[反対称](フェルミ粒子)かのどちらかであるということを,量子力学の原理として証明抜きで導入します。その結果,この要件を満たす波動関数を規格化(確率解釈するため)する過程で波動関数を √n! で割ってやる必要が生じ,その波動関数を用いて計算すれば,古典論 (熱力学や古典統計力学) で必要となる 「場当たり的な対応なし」 にいつでも直接,混合エントロピーが計算できるようになります。)